Currently, the genetic component of Rheumatoid Arthritis (RA) is not entirely described despite the identification of the HLA-DRB1 gene (and specifically the Shared Epitope (SE) alleles) and more than 100 susceptible genes.
One possible explanation of this missing heritability is that rare or low-frequency variants may also contribute to the underlying genetic risk. Advances in sequencing technologies (Next Generation Sequencing - NGS) now allow testing the hypothesis “rare variant-common disease”. If the identification of rare variants in cases-controls studies is challenging, it can be improve with familial data where an aggregation of theses variants can be observed.
NGS data can also be exploited to explore other causes of “missing heritability” such as Copy Number Variation (CNV) and Gene-Gene (GxG) interaction in a particular biological pathway.
In order to investigate these different causes of missing heritability, we benefit of familial samples collected through the ECRAF consortium (European Consortium for Rheumatoid Arthritis Families). All our sequencing data have been/are produced by the Centre National de Recherche en Génomique Humaine (CNRGH/ Institut François Jacob, CEA, Evry) with an Illumina platform.
Characterization of new rare variants (SNV & indels)
This project has been initiated with the search for rare variants (Single Nucleotide Variants (SNV) or indels) from whole exome data in French RA-multiplex families where HLA-DRB1 SE alleles segregate (HLA-DRB1+) since clinical heterogeneity has been observed for HLA-DRB1+ cases. The calling and annotation of variants led to categorise them according to their MAF (Minor Allele Frequency), their predicted effect on proteins (cADD score), and penetrance and phenocopy values.
Variants with a MAF <1%, a cADD score ≥ 30, a full penetrance and no phenocopy were first analysed with the p-VAAST software that allows to compute a global score based on a linkage score in our families and an association score by comparing the genetic information of affected subjects of our families to external controls (chosen with European ancestor origin and sequenced with a similar Illumina platform). After validation, a nonsense variant, introducing a premature stop codon at the beginning of the SUPT20H gene, has been identified. This gene is involved in the regulation of macroautophagy, which plays a key role in the pathogenesis of RA (DOI : 10.1371/journal.pone.0213387).
In addition, the analysis of whole genome sequencing data in French RA-multiplex families where no HLA-DRB1 SE alleles segregate (HLA-DRB1-) is ongoing to identify potential causal variants that could be specific of non HLA-DRB1 genetic component but also to determine if there is a similar part of the genetic component.
Characterization of CNV 
The study of CNV (Copy Number Variation) has already be initiated in the lab in order to evaluate the association of CNV candidate gene in our familial sample (see Copy Number Variants part). However, all the produced NGS data will be also exploited in order to identify new CNV in RA. Before to adapt the bio-informatics pipelines developed for SNVs and indels, simulation studies are ongoing in order to choose the better algorithm to determine CNV from whole exome data in our HLA-DRB1+ samples. This will be pursued for HLA-DRB1- samples. If the advantage of whole genome sequencing is that it can capture the CNV breakpoints outside the exons, the reliability of different tools will also be evaluated.
The new characterized CNVs will be validated in additional sample and compared to an updated human CNVs map since its last version dates from 2015. This map has been constructed in order to compile the CNVs detected on healthy individuals from various origins, and harvested on the Database of Genomic Variants (DGV). An in-house pipeline has been developed to update the 2015 CNV map and to estimate the CNV event frequencies (gain, loss, gain+loss). Our results will also be technically validated by digital PCR methodology developed in the lab (see Copy Number Variants part).
Pathways and interaction GxG
For genes carrying variants with incomplete penetrance identified by the preceding analyses of NGS data, over-representation (ORA) or gene enrichment (GSEA) analyses has been performed in order to identify biological pathways. If the GSEA approach has been developed for expression data, some adaptations have been made for SNP data.
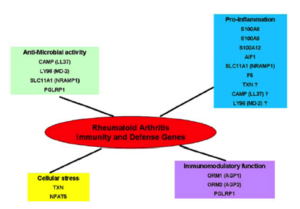
In the characterized biological pathways, interactions GxG is evaluated with multifactor dimensionality reduction approaches adapted for logistic regression. The biological impact of our results could then be evaluated through an interactive molecular map for RA that is currently developed in the lab through a system biology approach (see computational systems biology part). This map that consists in the construction of all pathways implicated in the disease, will allow to place genes in their functional context and to test their effects on the disease development.
The identification of new susceptible loci through NGS data would allow a better understanding of rheumatoid arthritis mechanisms, and would help to characterize new therapeutic targets or biomarkers useful for early diagnosis.